Xenium Ranger pipelines run on Linux systems that meet the minimum requirements listed below.
Requirements for rename subcommand:
- 8-core processor
- 4GB RAM
Requirements for relabel, resegment, and import-segmentation:
- 8-core Intel or AMD processor (32 cores recommended) with
avx2 - 64GB RAM (128GB recommended)
- 1TB free disk space
- 64-bit CentOS 7 and later/RedHat 7.4 or Ubuntu 16.04 and later; See the 10x Genomics OS Support page for details.
Running the following tissue area and input data have higher minimum memory requirements:
| Tissue area | Transcript counts | Subcommand | Minimum memory |
|---|---|---|---|
| 1cm2 dataset (e.g., full coronal mouse brain) | 500 million transcripts | Import transcript-assignment and viz-polygons (e.g., from Baysor) | 256GB RAM |
| 2cm2 dataset | 1 billion transcripts | Relabel | 256GB RAM |
| 2cm2 dataset | 1 billion transcripts | Import nucleus and cell boundary (e.g., GeoJSON from QuPath) | 488GB RAM |
| 2cm2 dataset | 1 billion transcripts | Import nucleus and cell label mask (e.g., from Cellpose) | 256GB RAM |
The pipeline can also run on clusters that meet these additional minimum requirements:
- Shared filesystem (e.g. NFS)
- Slurm batch scheduling system
- Xenium Ranger runs with
--jobmode=localby default, using 90% of available memory and all available cores. To restrict resource usage, please see the--localcoresand--localmemarguments forxeniumrangersubcommands. - In-house Xenium Ranger testing uses the following settings and we recommend customers do the same:
--localcores=32and--localmem=128. - Many Linux systems have default user limits (ulimits) for maximum open files and maximum user processes as low as 1024 or 4096. Because Xenium Ranger spawns multiple processes per core, jobs that use a large number of cores can exceed these limits. 10x Genomics recommends higher limits.
| Limit | Recommendation |
|---|---|
| user open files | 16k |
| system max files | 10k per GB RAM available to Xenium Ranger |
| user processes | 64 per core available to Xenium Ranger |
There are three primary ways to run Xenium Ranger:
- Single server: Xenium Ranger can run directly on a dedicated server. This is the most straightforward approach and the easiest to troubleshoot. The majority of the information on this website uses the single server approach.
- Job submission mode: Xenium Ranger can run using a single node on the cluster. Less cluster coordination is required since all work is done on the same node. This method works well even with job schedulers that are not officially supported.
- Cluster mode: Xenium Ranger can run using multiple nodes on the cluster. This method provides high performance, but is difficult to troubleshoot since cluster setup varies among institutions.
Recommendations and requirements, in order of computational speed (left to right):
| Cluster Mode | Job Submission Mode | Single Server | |
|---|---|---|---|
| Recommended for | Organizations using an HPC with SLURM for job scheduling | Organizations using an HPC | Users without access to an HPC |
| Compute details | Splits each analysis across multiple compute nodes to decrease run time | Runs each analysis on a single compute node | Runs each analysis directly on a dedicated server |
| Requirements | HPC with SLURM for job scheduling | HPC with most job schedulers | Linux computer with minimum 8 cores & 64 GB RAM (but see above) |
These plots are based on time trials using Amazon EC2 instances and Xenium Ranger v3.0.0.
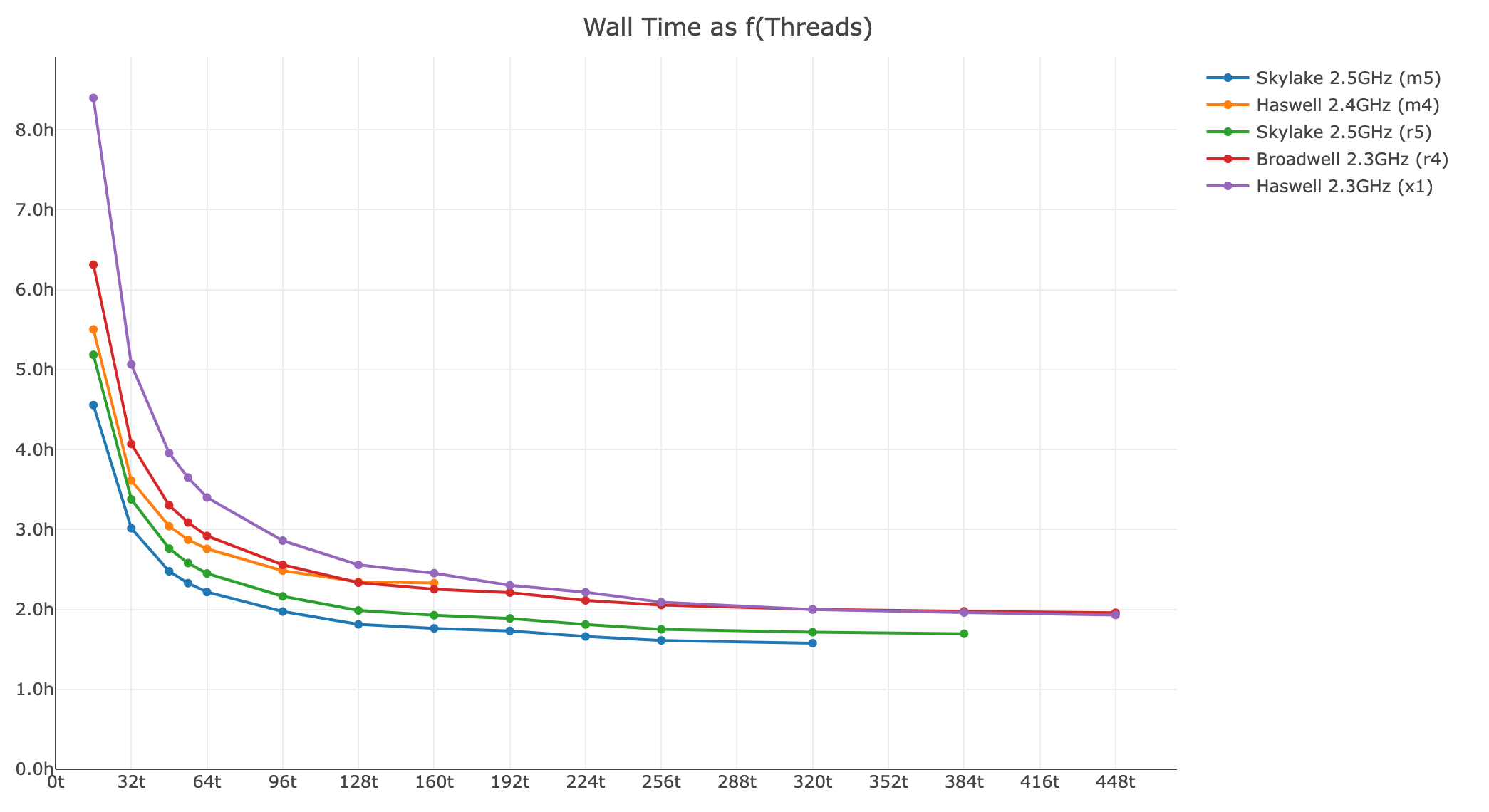
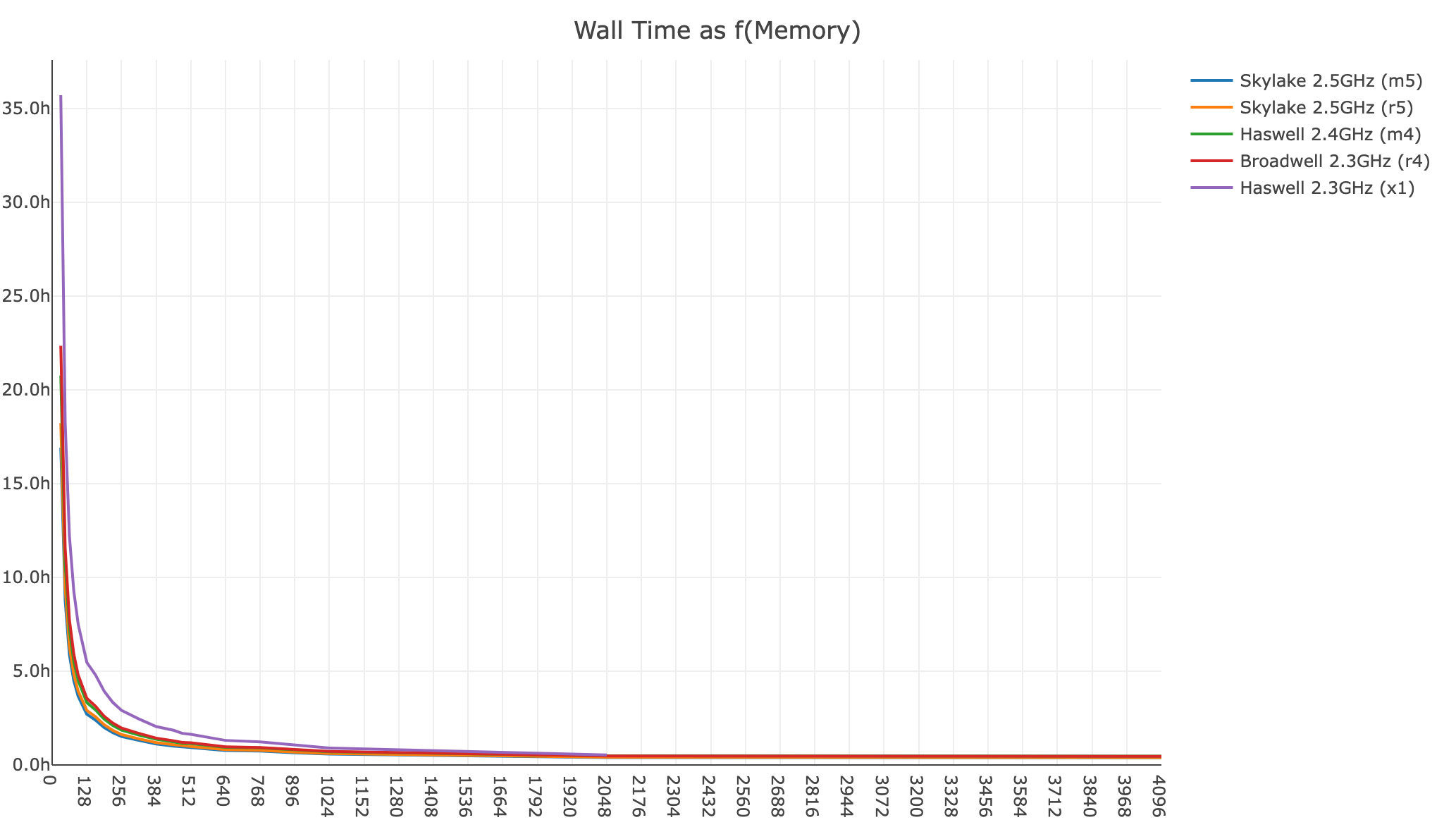
Shown below is a xeniumranger resegment analysis for a 140 FOV Xenium In Situ Gene Expression dataset (77,144,011 transcripts):
- Wall time as a function of memory
- Wall time as a function of threads
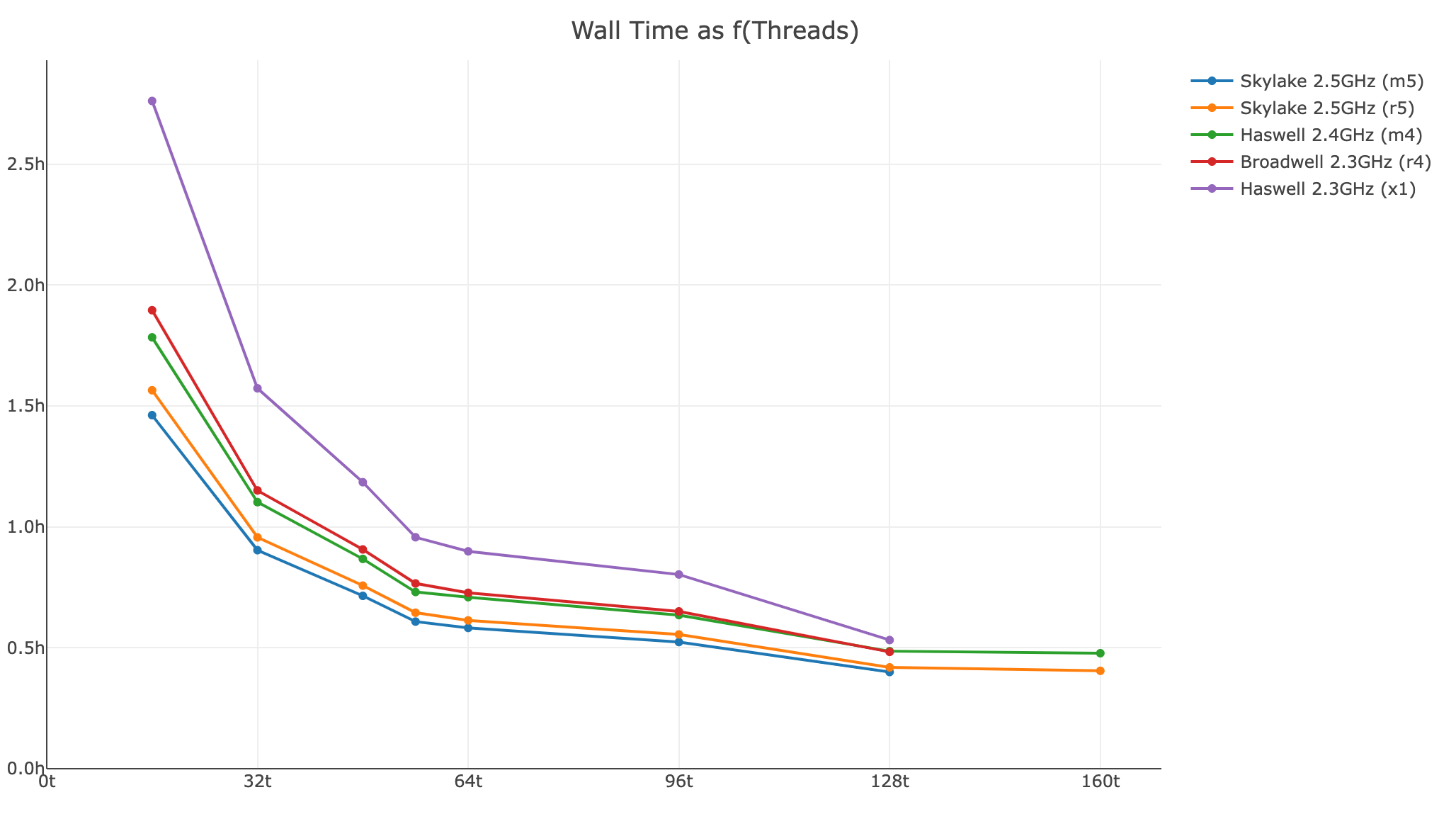
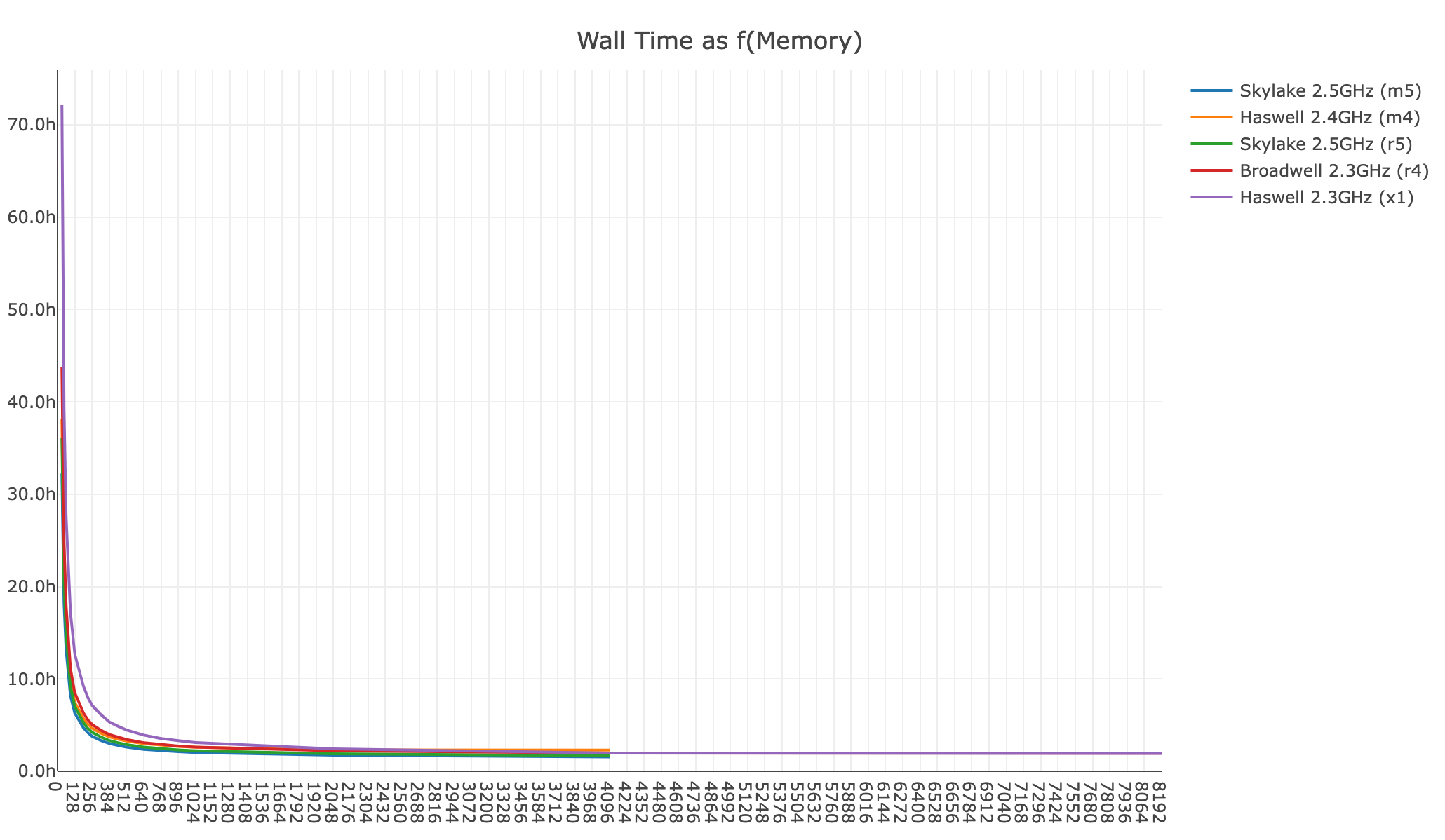
Shown below is a xeniumranger resegment analysis for a 200 FOV Xenium Prime 5K In Situ Gene Expression with Cell Segmentation Staining dataset (514,266,035 transcripts):
- Wall time as a function of memory
- Wall time as a function of threads