Spatially defined ligand/receptors as potential novel biomarkers for response to immunotherapy

The challenges that immuno-oncology researchers face can boil down to one thing: resolving tumor heterogeneity. And the answer to these challenges lies in better biomarkers.
However, biomarkers themselves are heterogeneous. While a biomarker is typically defined as the presence or absence of something—a gene, an analyte, a protein marker—they encompass so much more than that.
Xenium In Situ single cell spatial imaging is helping researchers realize that spatial information is not only important for a greater understanding of immuno-oncology, but it can be a biomarker in and of itself. Where genes are expressed, the pattern they're expressed in, how cells are positioned relative to one another, how they communicate—all these factors contribute to the heterogeneity of tumors which, in turn, makes these spatial contexts potential biomarkers of tumor-immune response, treatment response, and more.
Researchers from Fynn Biotechnologies realized these possibilities and, in a recent preprint (1), sought to characterize tumor heterogeneity and identify biomarkers of treatment response using Xenium In Situ. By leveraging massively multiplexed single cell spatial transcriptomics to analyze a variety of breast cancer subtypes with varying immunotherapy treatment status, these researchers were able to:
- Resolve the cellular composition of the tumor microenvironment and dissect its spatial organization
- Combine analyses of cellular composition with spatial colocalization to provide a better discrimination of the TME in breast cancer subtypes
- Identify the cellular and molecular architecture that defines treatment response to immunotherapy in diverse breast cancer subtypes
- Leverage spatially defined ligand/receptors as potential novel biomarkers for response to immunotherapy
Breaking down the cellular composition of breast cancer subtypes and validating correspondence of RNA with IHC data
Even among cancers, breast cancer is extraordinarily heterogeneous. The researchers used the Xenium Human Breast Panel to transcriptionally characterize a pathologically and biologically diverse set of four breast cancer samples, consisting of:
- Two luminal carcinomas, one with chest wall metastasis (LBC-Chest-Met) and one with lung metastasis (LBC-Lung-Met)
- One lymph node metastasis from a carcinoma positive for HER2 and negative for HR (HER2+/HR-LN-Met)
- One lymph node metastasis from triple-negative carcinoma (TNBC-LN-Met)
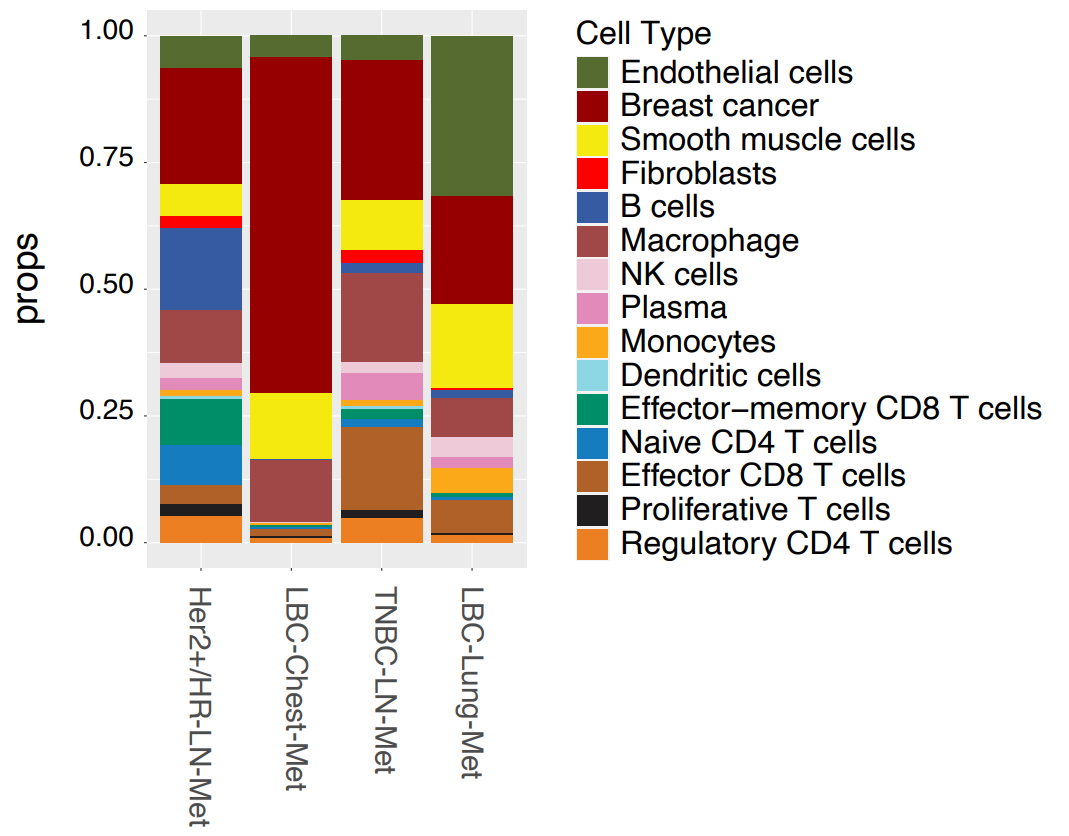
Using the gathered gene expression data, cell types were annotated based on marker genes and subdivided by major cell types, immune cell types, and T cell types. 15 cell types were initially resolved across all samples, which further segregated into expected hierarchical lineages (Figure 1). Immune cells split into myeloid and lymphoid clusters, with the latter dividing into various T-cell subtypes, including effector, regulatory, naive, and natural killer T cells.
Breast cancer subtypes showed distinct cell compositions. Samples from lymph node metastasis (HER2+/HR-LN-Met and TNBC-LN-Met) exhibited similar cell compositions, while luminal carcinoma samples differed markedly. LBC-Chest-Met showed a much higher tumor content than any other sample, while LBC-Lung-Met contained high proportions of smooth muscle and endothelial cells.
This initial data provided single cell information, but the true strength of Xenium lies in combining this with spatial context. Researchers overlaid cellular localization data on H&E stain images, generating cell maps that matched well with anatomical features (Figure 1).
Finally, one of the challenges of using gene expression data is that RNA levels do not necessarily correspond to protein levels. To further confirm the validity of their data, the group used IHC to compare RNA and protein expression levels of four well-established histological markers (ESR1, PGR, ERBB2 (Her2), and MKI67 (Ki67)) and demonstrated an overall good correspondence between RNA and protein data across breast cancer subtypes. Extrapolating these findings, high-plex transcriptomics approaches could be a way to provide orders of magnitude more data, more easily, from limited and precious patient samples than legacy methods.
Combining cellular composition with spatial colocalization provides a better discrimination of the TME in breast cancer subtypes
The authors then looked at the cellular composition and organization of both tumor-enriched and immune-stroma regions of interest (ROIs) in HER2+/HR-LN-Met,TNBC-LN-Met, and LBC-Chest-Met.
Focusing first on tumor-enriched ROIs, they found that LBC-Chest-Met ROIs exhibited extreme enrichment of tumor cells compared to HER2+/HR-LN-Met and TNBC-LN-Met. Interestingly, the latter two metastasis samples exhibited extensive infiltration of a variety of immune and T cell types.
ROI analysis of immune-stroma regions divided the cancers into two groups: Her2+/HR-LN-Met (which presented with predominant B cells and CD8 T cells) and LBC-Chest-Met and TNBC-LN-Met (where smooth muscle, endothelial cells, and macrophages were the major cell types) (Figure 2). Underscoring the heterogeneity of these cancers and the importance of spatial context, the spatial distribution of these cell types in LBC-Chest-Met and TNBC-LN-Met differed markedly.
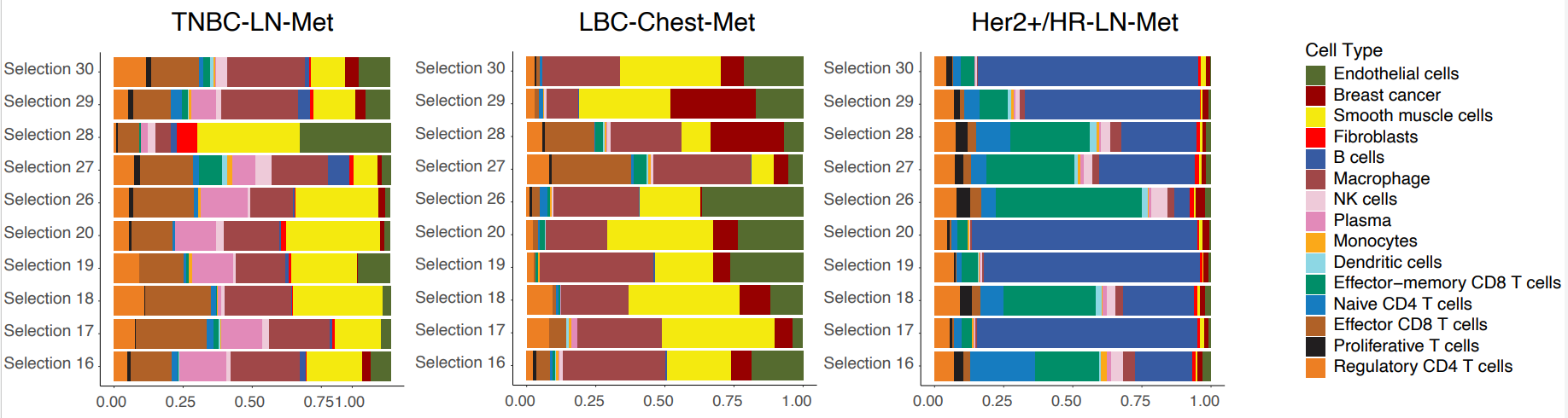
Next, the authors focused on cellular colocalization. In spite of some similarities in cell composition, each breast cancer subtype showed a unique TME. For instance, LBC-Lung-Met showed a strong colocalization of immune cells and macrophages with CD8 T cells, while LBC-Chest-Met showed cancer cells largely interacting with B cells.
Taken together, this data highlights not just the heterogeneity of cell composition in cancer, but the importance of understanding how those cell types interact to better understand the TME in disparate types of breast cancer.
Cellular and molecular architecture defines treatment response in diverse breast cancer subtypes
How immune cells are organized in tumors may influence how effectively anti-cancer therapeutics, such as immune checkpoint inhibitors (ICIs), work. To get at how this may occur, the authors looked at patients who’d received anti-PD-1 treatments and compared the TMEs of responders versus non-responders.
Using a now familiar strategy, the researchers started at the cellular level, then drilled down into the spatial organization of the TME. At the cell level, non-responders showed a higher proportion of cancer cells; in contrast, and consistent with anti-tumor response, responders showed increased proportions of immune cells (T cells, plasma cells, monocytes, and macrophages).
To look at spatial organization of the TME, the authors generated cellular neighborhoods (CNs) across each tissue sample by categorizing the 10 cells nearest to each individual cell, then compared the proportion of each CN in responders versus non-responders. Consistent with expectations, the CNs most enriched in responders were those linked to anti-tumor activity, and included both cytotoxic T-cell and T-cell/tumor-interacting macrophage neighborhoods (Figure 3).
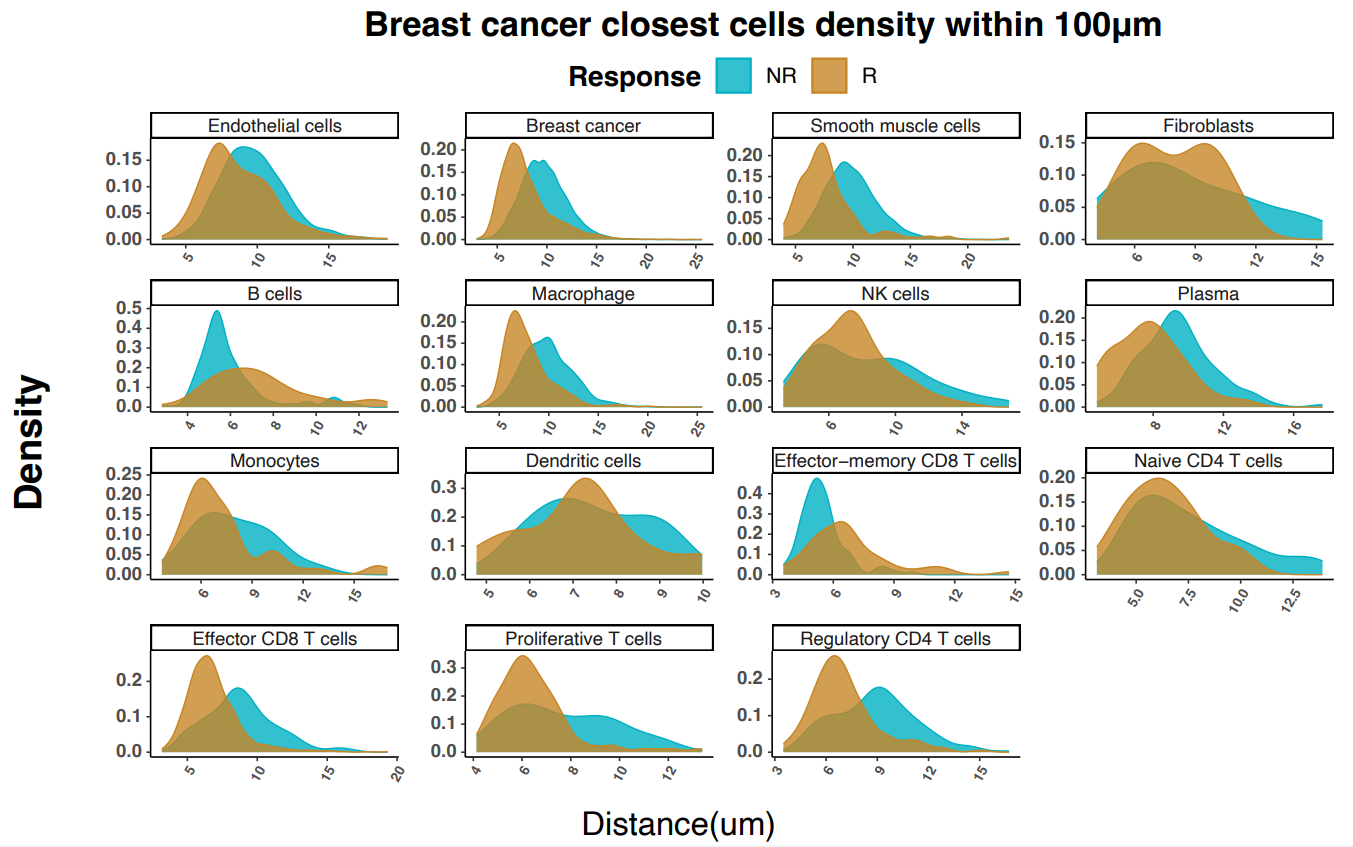
Importantly, the authors noted that the cytotoxic T-cell and tumor-interacting macrophage neighborhood (CN5) was more prevalent and tended to be located closer to tumors in the responder group. Zooming in even further, they found that a relatively rare Ki-67+ proliferative T-cell subtype was disproportionately represented close to tumors in responders versus non responders. Combined with a responder-specific co-recruitment of proliferative T cells, effector T cells, and macrophages, these findings shed light on how unifying cellular and spatial data can provide better insights into treatment responses in diverse breast cancer subtypes.
Spatially defined ligand/receptors as potential biomarkers
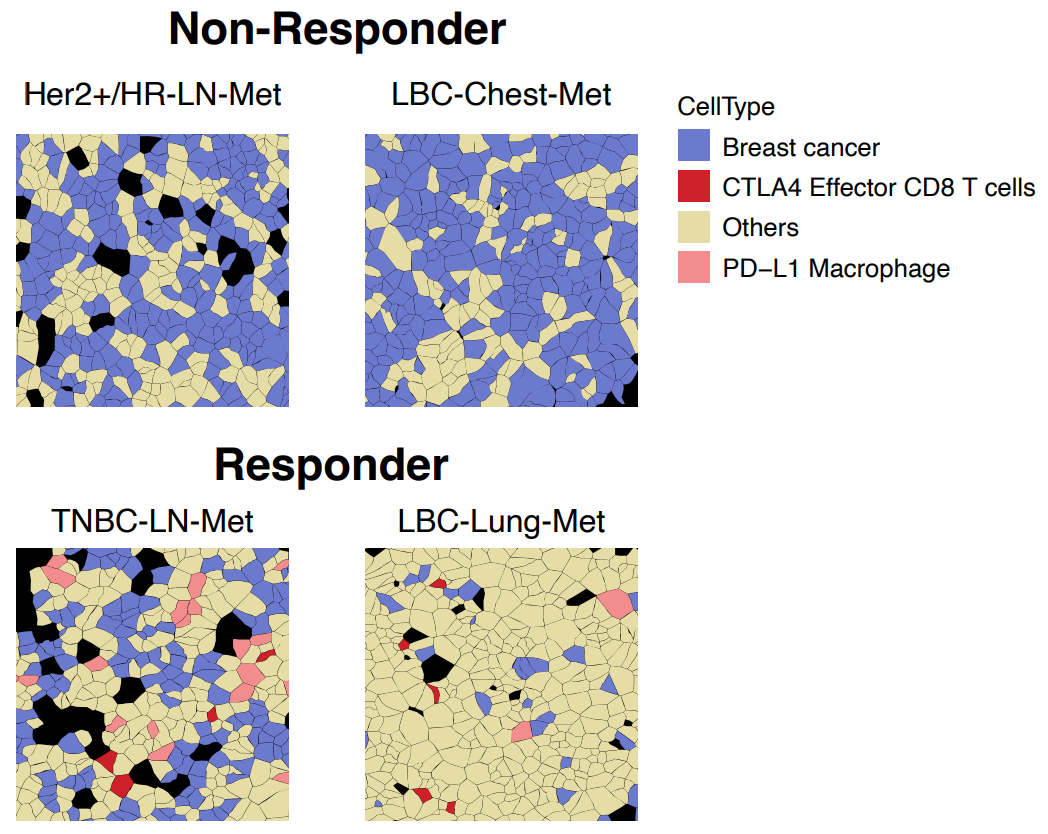
As altered cell–cell communication represents a potential mechanism underlying different responses in responders versus non-responders, the researchers next looked at ligand–receptor pairs in tumor-enriched ROIs.
Some pairings, such as CD86/CTLA4 (T-cell-inhibitory macrophage and T-cell communication), were seen in both responders and non-responders. However, T-cell-stimulating CD274/CD80 communication on DCs, T cells, and macrophages was only seen in responders. Again underscoring the heterogeneity of breast cancer subtypes, this CD274/CD80 interaction was also seen occurring between CD8-positive T cells and other immune cells types in one TNBC-LN-Met responder. One of the most intriguing findings from this was that, “Consistently, the spatial cell annotation from Xenium data demonstrated that the infiltrated CTLA4-positve effector T cells and PD-L1-positive macrophages directly communicate with the cancer cells exclusively in the responders” (Figure 4).
Finally, the researchers sought to leverage their findings for potential biomarker discovery. They looked at the expression of the known ICI biomarker PD-L1 (CD274) RNA in tumor cells, as well as RNA levels of other potential biomarkers, including PD-L1 (CD274), PD-L2 (PDCD1LG2), NKG7, and CD80. Not only were all of these RNAs significantly upregulated in responders, but they all exhibited greater statistical significance than PD-L1, raising the intriguing possibility of using single cell spatial for further exploration and validation of potential biomarkers.
Forging the future with single cell spatial
These findings from Fynn Biotechnologies highlight the importance of understanding both cellular and spatial heterogeneity in immuno-oncology. By showing the complexities of how the TME differs in cellular composition, colocalization, and communication—all of which require both single cell and spatial data to resolve—their preprint sheds light on how the molecular and spatial architecture of various types of breast cancer can vary, raising the chance of new biomarkers and laying the groundwork for new possibilities in patient stratification.
Interested in how Xenium can accelerate your immuno-oncology work? Explore the Xenium Immuno-Oncology Panel and other assays, or expand your analysis to ultra high-plex with Xenium Prime 5K.
References:
- Wang N, et al. Spatial single-cell transcriptomic analysis in breast cancer reveals potential biomarkers for PD1 blockade therapy. Research Square (preprint) (2024). doi: 10.21203/rs.3.rs-4376986/v2
Blog author:
