Organoids bring drug discovery and development to the culture hood

Non-human animal models are usually the first to try new and developing therapeutics. For more than 50 years, the United States Federal Drug Administration (FDA) required all drugs be tested in a rodent and non-rodent animal prior to human clinical trials. But in December 2022, the FDA waived that requirement. Now, drugs thoroughly tested using either animal or non-animal models—including organs-on-a-chip, organoids, and computer models—can proceed to clinical trials, upon FDA approval (1).
The FDA also requested an additional $5 million in their annual budget to support a new, FDA-wide New Alternative Methods Program to “reduce animal testing through the development of qualified alternative methods and spur the adoption of methods for regulatory use that can replace, reduce, and refine animal testing (2).”
The FDA is gearing up for the coming revolution of human cell–derived drug discovery and preclinical models. Although these models are still in their infancy, growing evidence suggests they could help not only decrease the excessive cost and time spent on preclinical research, but increase success during clinical trials—9 out of 10 clinical trials fail due to low drug efficacy or unexpected toxicities (3).
Organoids, in particular, have emerged as a promising new model for drug discovery and development. Organoids are self-organized three-dimensional tissue cultures derived from stem cells, and can match the complexity of an organ, or express certain aspects of the organ. Human cell–derived organoids can catch toxicities mice miss. For example, human “gastruloids” better model fetal toxicity caused by thalidomide—a drug that causes birth defects in humans but not mice—than mouse gastruloids (4). Organoids derived from patient cells enable researchers to identify human-specific drug targets and disease mechanisms. Researchers can even develop patient-specific organoids to determine the best treatment for an individual patient.
Single cell RNA sequencing (scRNA-seq) helps translational researchers use organoids during the drug discovery and development process. Researchers can validate that their organoids are truly representative of human tissue diversity and observe transcriptomic differences between diseased and non-diseased organoids or organoids derived from patient cells before or after treatment.
In this post, we highlight four papers that employed Chromium Single Cell Gene Expression technology and organoids in their efforts to identify druggable disease pathways, screen drugs, model drug metabolism, and explore patient-specific therapeutic responses.
Population screening for genetic susceptibility
Nonalcoholic fatty liver disease (NAFLD)—the most common form of liver disease impacting billions of people worldwide—is a condition where fat builds up in the liver. In some cases, NAFLD progresses to nonalcoholic steatohepatitis (NASH), resulting in liver inflammation and damage. Although there are known genetic risk factors and pre-existing conditions, including diabetes and obesity, that increase an individual's susceptibility to developing NAFLD/NASH, these factors aren’t consistent. With no current treatments—aside from lifestyle changes—available for either condition, an improved understanding of the genetic underpinnings and risk factors involved are needed to develop therapeutic and prophylactic treatments.
Researchers from Tokyo Medical and Dental University and Cincinnati Children’s Hospital Medical Center (CCHMC) identified a specific genetic mutation in glucokinase regulatory protein (GRP) that increases a patient’s risk of developing NASH if they had diabetes, but decreases a non-diabetic patient’s risk of developing the disease (5).
To reveal the complexities of GRP as a NASH risk factor, the researchers first established a “population organoid panel (PoP)” to screen for NASH phenotype–genotype associations. They reprogrammed cells from 24 donors into induced pluripotent stem cells (iPSCs), and differentiated them into foregut progenitor cells. They embedded clusters of foregut progenitor cells from each donor into a large, basement-membrane matrix gel to generate 24 individual human liver organoids (HLOs). They conducted scRNA-seq using our Chromium Single Cell 3’ Gene Expression technology to validate that the embedded PoP organoids contained three key cell types similar to those found in the liver—hepatocyte-like, macrophage-like, and stellate-like cells.
They treated the PoPs like the liver is treated in someone with NAFLD/NASH by packing their media with extra fatty acids. Human liver organoids developed from patient samples with two copies of the specific GRP variant took up fat more effectively than organoids developed from other patients.
When the researchers retrospectively analyzed genetic data from NASH patients, they discovered that patients with diabetes and two copies of the GRP mutation had high levels of liver inflammation while patients without copies of the mutation had low levels of inflammation. Their findings seem to be clinically significant as well. The presence, or lack thereof, of the mutation affected how well patients responded to commonly used diabetes treatments, including metformin.
“This finding suggests a new treatment approach that could make a real impact in the lives of diabetic patients with the mutation who do not respond to standard medication,” said Masaki Kimura, PhD, first author of the study and a research associate from CCHMC (6).
Mini-kidneys, major discoveries
Researchers from the University of Southern California (USC) recently developed a scalable organoid platform to generate thousands of kidney organoids, which they used to screen 247 enzyme inhibitors for effectiveness against a mutation found in people with autosomal dominant polycystic kidney disease (ADPKD) (7).
The 8 million people worldwide with ADPKD develop large, fluid-filled cysts in their kidneys that slowly render them non-functional, causing a variety of secondary life-threatening conditions, including bone disorders, cardiovascular issues, and anemia. But the only FDA-approved drug on the market only slows its progression in a subset of patients.
The USC researchers decided to help identify new drugs by developing iPSC-derived organoids. They first differentiated iPSCs into kidney organoids for 28 days in plates containing 100s of microwells that collected clusters of cells. By day 14 of the differentiation, they observed kidney-specialized cells, and, by day 25, they saw one or two nephron-like structures—the kidney’s filtering units—per organoid.
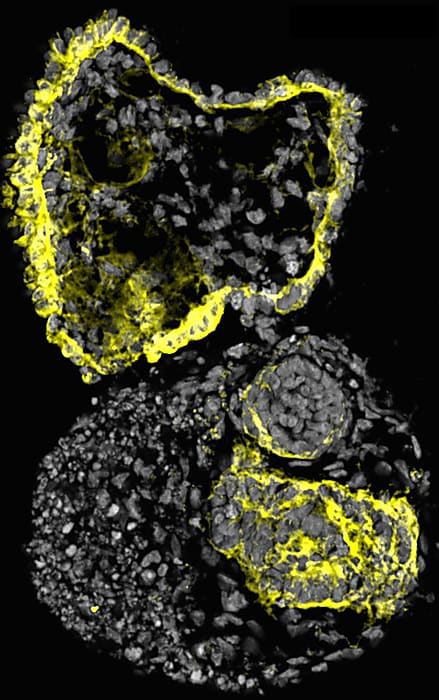
They used Chromium Single Cell 3’ Gene Expression technology to analyze the transcriptional profiles of these organoids during days 10, 14, 16, and 28. They assigned cluster identities using the expression of well-established cell markers with Seurat version 3. Their analysis mirrored scRNA-seq analyses from other groups including transcriptional signatures of a variety of nephron progenitor cells (NPCs) and non-nephron cells, such as intestinal cells.
They dived deeper into their data by subsetting cells annotated to nephron-related lineages to develop a nephron development trajectory. Their final extraction contained 8,654 cells from four time points (days 8, 14, 16, and 18). They found nephron progenitor-like clusters in day-8 samples, while later time points revealed more specialized kidney cells, including podocyte-like cells and proximal tubule-like cells. Most importantly, transcription factors that are known to be present in early human kidneys were expressed in their mini-kidneys—including EYA1, MEOX1, SIX1, and SIX2—indicating that the core nephron developmental program was likely activated during differentiation.
They used scRNA-seq to relieve any doubt their organoids weren’t on par with in vivo models. They validated scRNA profiles from two batches of their own organoids from multiple time points to ensure reproducibility. Then, they compared their organoid differentiated cells to 4,405 nephrogenic cells from week-17 human fetal kidney datasets, as well as to other kidney organoid datasets produced by other researchers. The core transcription factors correlated across all datasets they compared, but there were some transcription factors involved in later stages of maturation expressed in the human fetal kidney that were not expressed in their organoids.
But the organoids did mature when the researchers implanted them into mice. The nephron-like structures developed vasculatures and could even filter waste. Overall, they showed the organoids had most of the cells and developmental programs needed to build a kidney.
To put their model to the test, they used CRISPR editing to inactivate pyruvate dehydrogenase kinase 1 (PDK1) or 2 (PDK2)—the mutation responsible for over 90% of all ADPKD cases—in the kidney organoids and treated them with 247 different protein kinase inhibitors. Nine of those inhibitors inhibited the growth of cysts without stunting the growth of the organoid. Quinazoline was particularly effective, inhibiting cyst development in organoids carrying either PDK1 or PDK2 mutations. This is promising, given that several quinazoline-derivatives are FDA-approved to treat certain forms of cancer.
“Our organoids proved to be very useful for identifying therapeutic drug candidates that merit further study for the treatment of ADPKD,” said Chen (Jack) Song, PhD, an author on the study and a postdoctoral Amgen scholar at the University of Southern California in a press release (8).
Characterizing drug metabolism in little livers
Drugs act on more than the targeted diseased organ or tissue. In fact, most drugs are inactive until they are metabolized—their metabolites are usually responsible for a drug’s efficacy and toxicity. The liver—the primary site of drug metabolism—is a critical component of drug discovery and preclinical testing. But testing with mice has been challenging because the main family of drug-metabolizing enzymes, cytochrome P450 (CYP450) enzymes, vary significantly between mice and humans. Mice have 72 functional CYP450 genes, while humans only possess 27 (9). Several in vitro models are available for drug metabolism assays, but none offer a clear solution.
Recently, a group of researchers developed human hepatic organoids (hHO) with high CYP450 expression and activity that recapitulated the metabolism and CYP450-mediated drug toxicity of previously characterized compounds—even more effectively than in established cell lines, such as primary human hepatocytes (PHHs) (10).
Before putting their hHOs to the test though, they first used Chromium Single Cell 3’ Gene Expression to validate that their organoids contained the expected cell populations. They identified 19 cell clusters and, based on the gene expression patterns and gene ontology analysis, they defined three groups of cells with biliary-, hepatocyte-, and gallbladder-like features.
They integrated their data with a previously published scRNA-seq analysis of human tissues. The analysis revealed that the hHOs primarily expressed hepatocyte- and biliary-like cells, and a small portion of gallbladder-like cells. The hepatocyte- and biliary-like cells clustered with bile-duct-inhabiting cholangiocytes in the human tissue and almost exclusively expressed cell-type-specific markers, including SERPINA1 and KRT19, respectively. The gallbladder-like cells, however, broadly expressed hepatic, biliary, and gallbladder markers, making their classification less definitive.
To test the functionality of their hHOs, they generated hHOs from human embryonic stem cells carrying a Wilson’s disease–causing mutation in the ATPase copper transporting beta (ATP7B) gene and tested its response to the most commonly prescribed Wilson's disease drug, D-penicillamine. Wilson’s disease is a rare, heritable disorder where copper accumulates in the organs causing a variety of symptoms, including coordination problems, depression, and jaundice.
hHOs carrying the ATP7B mutation accumulated far more copper and incurred more cell death than wildtype hHOs when they were treated with high concentrations of copper chloride. When they treated the ATP7B-mutation-carrying hHOs with D-penicillamine, cell viability improved.
The authors were excited about their results. They stated in the publication that their platform “will have applications not only in academic research, but also in the pharmacological industry.”
Clinical precision oncology in a dish
Organoid modeling in translational research not only presents the opportunity to speed up and streamline discovery and development in the cell culture hood, it also has the potential to change treatment decisions by clinicians.
A team of researchers from the Terasaki Institute for Biomedical Innovation (TIBI) and Duke University, led by TIBI’s chief scientific officer and researcher, Xiling Shen, PhD, put the organoid’s capacity for making speedy precision medicine accessible on center stage in their 2022 study published in Cell Stem Cell.
The team developed a workflow that employs droplet emulsion microfluidics to generate thousands of “micro-organospheres” (MOSs) from precious oncology patient samples in an hour or less, enabling downstream drug testing. The researchers used this workflow to test potential drugs on MOSs derived from colorectal cancer samples taken from individuals in an active clinical study—they assessed drug responses within 14 days, a quick enough turnaround to realistically inform patient treatment decisions in the real world (11).
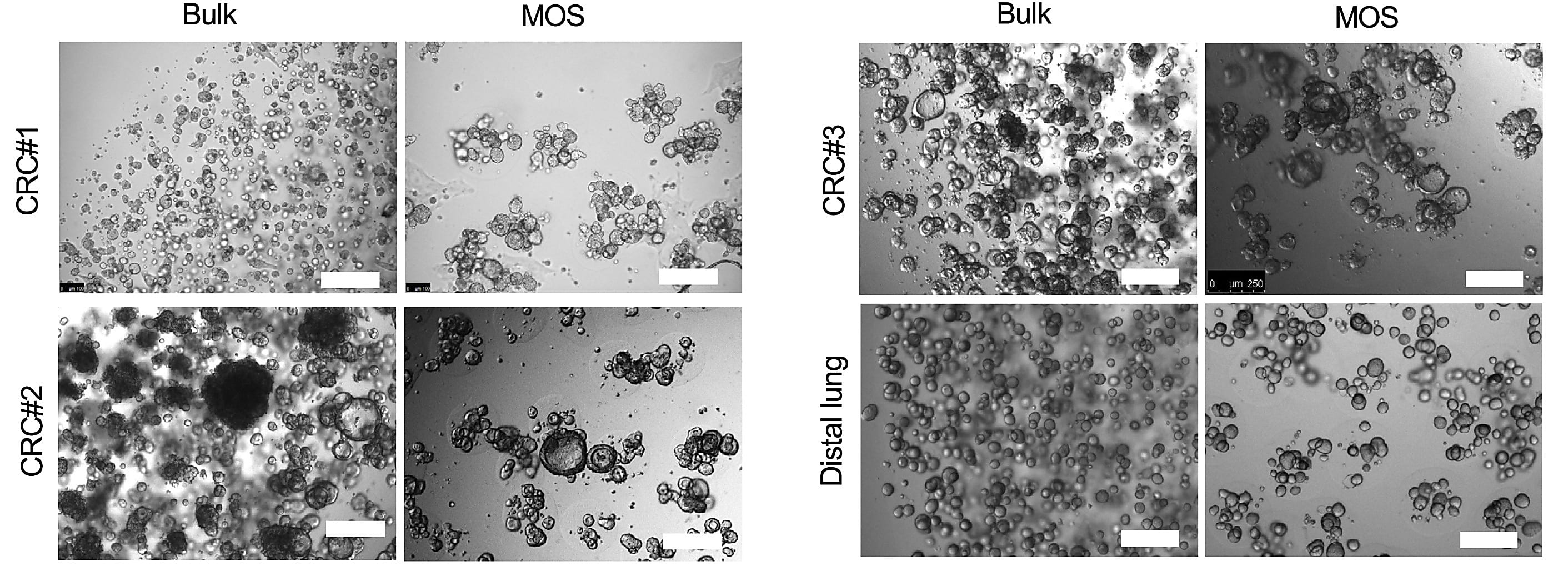
Detailed scRNA-seq analyses of the MOSs they collected with our Chromium Single Cell 3’ Gene Expression technology were critical in establishing their model and validating its functionality during drug testing.
They first used scRNA-seq to compare tumor and stromal cell populations between primary human tumor tissue and their MOS derivative in six pairs of matched patient tumor samples—three lung cancer samples, one kidney cancer sample, one ovarian cancer sample, and one CRC sample—and their MOS derivatives. Using uniform manifold approximation and projection (UMAP), they defined four general clusters across sample types—tumor cells, cancer-associated fibroblasts, and lymphoid or myeloid immune cells—and determined clustering and associated gene expression levels across each cell population was relatively consistent between primary tissues and their MOS derivatives.
They also employed UMAP visualization and cell-type labeling with the SingleR package to analyze cell-type-specific gene marker expression and label cell types. Critical markers for tumor cells, fibroblasts, myeloid cells, and lymphoid cells, including EPCAM, PDGFRA, LYZ, and CD3E, were consistent between primary tissue and MOS derivatives as well.
Overall, the authors concluded that MOSs recapitulate the stromal and immune components of the patient tumors in vivo, indicating they likely model common cancer therapy blockades, such as tumor microenvironment and fibroblast overgrowth.
The authors also used scRNA-seq to compare MOS derivatives before and after treatment with anti-cancer drugs. Specifically, they treated CRC MOSs with monoclonal antibody ESK1 (ESK-1 tumor binder, Roche proprietary CD3) to induce a cytotoxic T-cell response. Treatment did initiate apoptotic death, and scRNA-seq analysis post-treatment revealed that the abundance of all cell types was drastically reduced, indicating that treatment affected both tumor and stromal cells and speaking to the power of their pharmacological model.
“The technology developed here is a groundbreaking advancement in physiological modeling for solid tumor diseases and personalized medicine,” said Khademhosseini (13). “It is sure to have a highly significant impact in the clinic.”
Are organoids the wave of the future?
Researchers won’t be tossing non-human animal modeling from their drug discovery and development workflows anytime soon. But they are increasingly incorporating in vitro human models like organoids, as evidenced by the highlighted studies above.
Some, like Wendy Jarrett, CEO of Understanding Animal Research, an animal research advocacy group based in the United Kingdom, emphasize that organoid modeling is in its infancy and animal models are still needed to capture the full effects a drug could have on clinical trial participants. “We can drop a new [candidate drug] onto a bunch of liver cells. And we can see that it doesn’t damage them. But what we don’t know is whether it’s going to make the person cough, whether it’s going to damage their intestines or their brain,” she told Science News (1).
Despite this, the gradual shift to in vitro models spurred pre-emptive changes in the drug approval process by the FDA, a strong signal that both regulators and researchers want to at least complement their animal studies.
Stephen Grossman, a former deputy assistant secretary of health who advises companies on their FDA applications, told Science News that this shift in FDA regulation “provides a little additional authority. It says in law: ‘Congress is cool that these discussions are going on’” (1).
Learn more about how our Single Cell Gene Expression technology can potentially strengthen your drug discovery research.
References:
- Wadman M. FDA no longer needs to require animal tests before human drug trials. Science News (2023). doi: 10.1126/science.adg6264
- U.S. Food and Drug Administration. FDA Seeks $8.4 Billion to Further Investments in Critical Public Health Modernization, Core Food and Medical Product Safety Programs. (2022). https://www.fda.gov/news-events/press-announcements/fda-seeks-84-billion-further-investments-critical-public-health-modernization-core-food-and-medical
- Seyhan AA. Lost in translation: the valley of death across preclinical and clinical divide – identification of problems and overcoming obstacles. Transl Med Commun 4: 18 (2019). doi: 10.1186/s41231-019-0050-7
- Mantziou V, et al. In vitro teratogenicity testing using a 3D, embryo-like gastruloid system. Reprod Toxicol 105: 72–90 (2021). doi: 10.1016/j.reprotox.2021.08.003
- Kimura M, et al. En masse organoid phenotyping informs metabolic associated genetic susceptibility to NASH. Cell 185: 4216–4232 (2022). doi: 10.1016/j.cell.2022.09.031
- https://www.eurekalert.org/news-releases/971650
- Tran T, et al. A scalable organoid model of human autosomal dominant polycystic kidney disease for disease mechanism and drug discovery. Cell Stem Cell 29: 1083–1101 (2022). doi: 10.1016/j.stem.2022.06.005
- https://www.eurekalert.org/news-releases/957767
- Bissig KD, et al. P450-Humanized and human liver chimeric mouse models for studying xenobiotic metabolism and toxicity. Drug Metab Dispos 46: 1734–1744 (2018). doi: 10.1124/dmd.118.083303
- Kim H, et al. Development of human pluripotent stem cell-derived hepatic organoids as an alternative model for drug safety assessment. Biomaterials 286: 121575 (2022). doi: 10.1016/j.biomaterials.2022.121575
- Ding S, et al. Patient-derived micro-organospheres enable clinical precision oncology. Cell Stem Cell 29: 905–917 (2022). doi: 10.1016/j.stem.2022.04.006
- Wang Z, et al. Rapid tissue prototyping with micro-organospheres. Stem Cell Reports 17: 1959–1975 (2022). doi: 10.1016/j.stemcr.2022.07.016
- https://www.eurekalert.org/news-releases/951879
About the author:
